Introduction
Goal
The aim is to : get familiar with datasets produced by Next Generation Sequencing (NGS).
Topics:
- accessing data in GEO / SRA/ ENA databanks from an accession number in a publication
- searching for public datasets in GEO
- understanding the FASTQ format used for reads files
- Assess the overall quality of a FASTQ read file
Datasets description
For this training, we will use various ChIP-seq datasets. ChIP-seq is a functional genomics assay to find all the regions in a genome bound by a particular protein (e.g. a transcription factor).
- Bacteria: ChIP-seq dataset produced by Myers et al [PDF] involved in the regulation of gene expression under anaerobic conditions in bacteria. We will focus on one factor: FNR.
- Zebrafish: ChIP-seq produced by Paik et al [PDF] to study the factor Cdx4 during development. We will focus on H3K27ac in bud-stage embryo.
Download a dataset
Goal: Identify the dataset corresponding to a studied article and retrieve the data (reads as FASTQ file) corresponding to one experiment.
1 - Obtaining an identifier for a chosen dataset
NGS datasets are (usually) made freely accessible for other scientists, by depositing these datasets into specialized databanks.
Sequence Read Archive (SRA) located in USA hosted by NCBI, and its european equivalent European Nucleotide Archive located in England hosted by EBI both contains raw reads.
How many sequences and bases are currently contained in ENA ?
Check the latest news to find information on the last release.
Functional genomic datasets (transcriptomics, genome-wide binding such as ChIP-seq,...) are deposited in the databases Gene Expression Omnibus (GEO) or its European equivalent ArrayExpress.
How many experiments are stored in ArrayExpress ?
Check the "Data Content" section. Note that the raw reads are not stored in ArrayExpressed, but linked to ENA.
What is the difference between Genbank and SRA ?
Within an article of interest, search for a sentence mentioning the deposition of the data in a database (tip: automatically search in the pdf "GEO" or "SRA"). In the Bacteria dataset [PDF] , the following sentence can be found at the end of the Materials and Methods section:
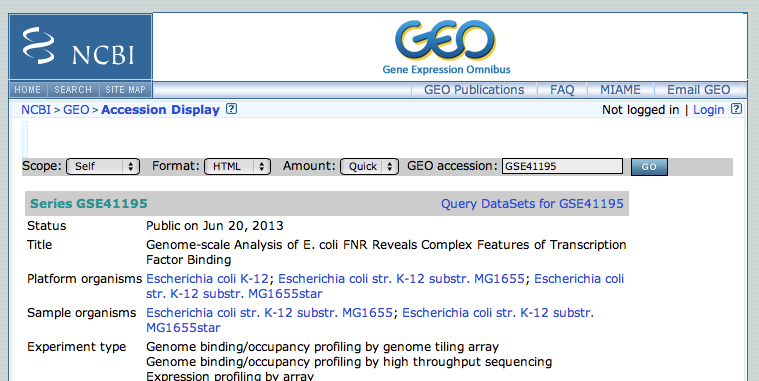
"All genome-wide data from this publication have been deposited in NCBI’s Gene Expression Omnibus (GSE41195)."
We will thus use the GSE41195 identifier to retrieve the dataset from the NCBI GEO (Gene Expression Omnibus) database.
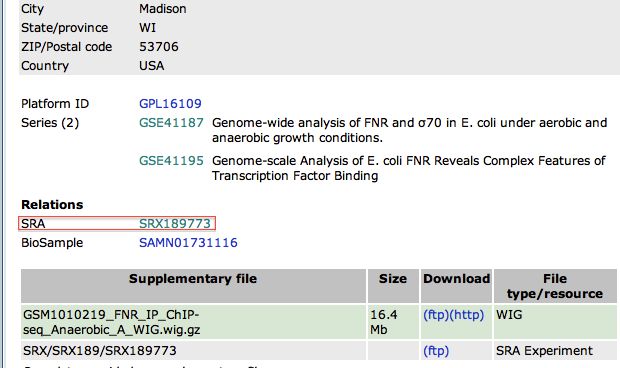
2 - Accessing GSE41195 from GEO
3 - Downloading FASTQ file from the EBI database
Although direct access to the SRA database at the NCBI is doable, in practice it's simpler and quicker to download datasets from the EBI (European Bioinformatics Institute) website, located in Europe. The EBI hosts the ENA database (European Nucleotide Archive), which encompasses the data from SRA.
- Go to the EBI website. Paste your identifier (SRX189773) and click on the button "search"
- Click on the first result. On the next page, there is a link to the FASTQ file. Download this file (= save this file on your computer). As FASTQ files are large, this will be the only sequence file that we download for this course.
- For the second article, redo all the steps above and find the link to the FASTQ file (without downloading it...)
At this point, you should know how to find a FASTQ file.
Searching GEO
Goal: Find datasets of interest (might not be published yet !)
1 - Searching the GEO
- Go to the NCBI website and select GEO datasets as the database to search.
- Search the term chip-seq
How many results were found ?
As of today, 11702 datasets in GEO match the term "chip-seq". This number keeps growing...
- The left part of the page aims as enabling filters on the current search. It's possible to filter on the organism, study type, tissue... Filter the results to obtain only zebrafish datasets.
What is the latin name for zebrafish ? How many results are found ?
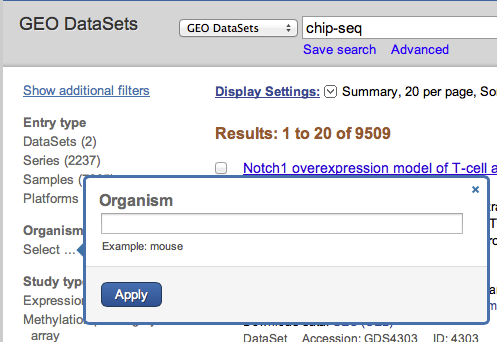
Under organism, click on the link Select.... By searching zebrafish, the system automatically proposes "Danio rerio", the latin name of this organism. This filter limits the search to 46 datasets.
2 - Analyze a GEO entry
- Click on the dataset "Zic3 interacts with distant regulatory elements to regulate zebrafish developmental genes".
When was this dataset released as public ?
What was the overall design ?
Is the dataset published (see Citation section) ?
On which sequencing platform was the data produced ?
On this example in zebrafish, the data was publicly released on Feb 27, 2014. This dataset is related to a publication in the journal Plos Genetics. The overall design is ChIP-seq targeting the protein Zic3 in wild type and sqet33 transgenic. Sequencing was performed on a Illumina Genome Analyzer, in Singapore.
3 - Search the GEO by keyword
- Go back to the search page, enter the term hox* and filter by organism zebrafish
How many datasets are available ?
44 datasets are related to one of the Hox gene family in zebrafish
- Go back to the search page, enter the term CLIP-Seq
Search in google what is CLIP-seq. How many CLIP-seq datasets were produced in Saccharomyces cerevisiae ?
Look at the top right of the page. 6 datasets related to CLIP-seq are counted.
At this point, you should know how to search the GEO database, filter the results, and read a GEO entry.
Viewing raw (FASTQ) data
Goal: view raw data in the terminal
1 - Obtain a FASTQ file
FASTQ files are generally larger than 1 Go. They cannot be opened by usual programs (and you should avoid trying opening them, as it could really freeze your computer). To view such big text files - text as in not binary files - you can use the terminal.
You should already have downloaded the Bacteria dataset FASTQ file on your computer. Do you remember where ?
If you haven't downloaded it yet, you can obtain it from the ENA database with accession SRX189773.
Save it on your Desktop.
2 - Viewing the FASTQ file
- Go to the Desktop folder
cd Desktop
- List the content of this folder. You should see the downloaded file SRR576933.fastq.gz. How large is the file ?
ls -lth
-rw-r--r-- 1 mthomas csb 121M avril 1 21:45 SRR576933.fastq.gz
- The file is zipped. Unzip the file
gunzip SRR576933.fastq.gz
- Check again the content of the folder How large is the fastq file ?
ls -lth
-rw-r--r-- 1 mthomas csb 453M avril 1 21:45 SRR576933.fastq
- View the 20 first lines (means the 5 first reads, as there are 4 lines per read)
head -n 20 SRR576933.fastq
Do you recognize the 5 reads in the FASTQ format ?
3 - Understanding the FASTQ file
- Use google to search for the specifications of the FASTQ format. Your FASTQ file is encoded in Sanger FASTQ.
Can you assess wether the first 5 reads are of good or bad quality ?
At this point, you should be able to view a FASTQC file in the terminal and recognize this format.
Quality control of the reads and statistics
Goal: Get some basic information on the data (read length, number of reads, global quality of dataset)
1 - Getting the FASTQC report
Before you analyze the data, it is crucial to check the quality of the data. We will use the standard tool for checking the quality of data generated on the Illumina platform: FASTQC.
- First check the help page of the program to see its usage and parameters.
fastqc --help
Check the description of the program. Do you understand what if does ? What is the input of this program ?
- Run fastqc program on your dataset.
fastqc SRR576933.fastq.gz
- Check the files output by the program.
ls
SRR576933_fastqc SRR576933_fastqc.zip SRR576933.fastq.gz
- Access the result.
cd SRR576933_fastqc
ls
fastqc_data.txt fastqc_report.html Icons Images summary.txt
- Open the HTML file fastqc_report.html in Firefox.
Analyze the result of the FASTQC program:
How many reads are present in the file ?
What is the read length ?
Is the overall quality good ?
Are there any concerns raised by the report ? If so, can you tell where the problem might come from ?
There are 3 603 544 reads of 36bp. The overall quality is good, although it drops at the last position, which is usual with Illumina sequencing, so this feature is not raising hard concerns. There are several "red lights" in the report. In particular, the per sequence GC content and the duplication level are problematic. If you check the "overrepresented sequences", you'll notice a high percentage of adapters (29% !). Ideally, we would remove these adapters (=trim) the reads, and then re-run FASTQC.
Results.
What are the adapters ?
2 - Organism length
Knowing your organism size is important to evaluate if your dataset has sufficient coverage to continue your analyses. For the human genome (3 Gb), we usually aim at the very least 10 Million reads for ChIP-seq.
- Go to the NCBI Genome website, and search for the organism Escherichia coli
- Click on the Escherichia coli str. K-12 substr. MG1655 to access statistics on this genome.
How long is the genome ?
Do both FASTQ files contain enough reads for a proper analysis ?
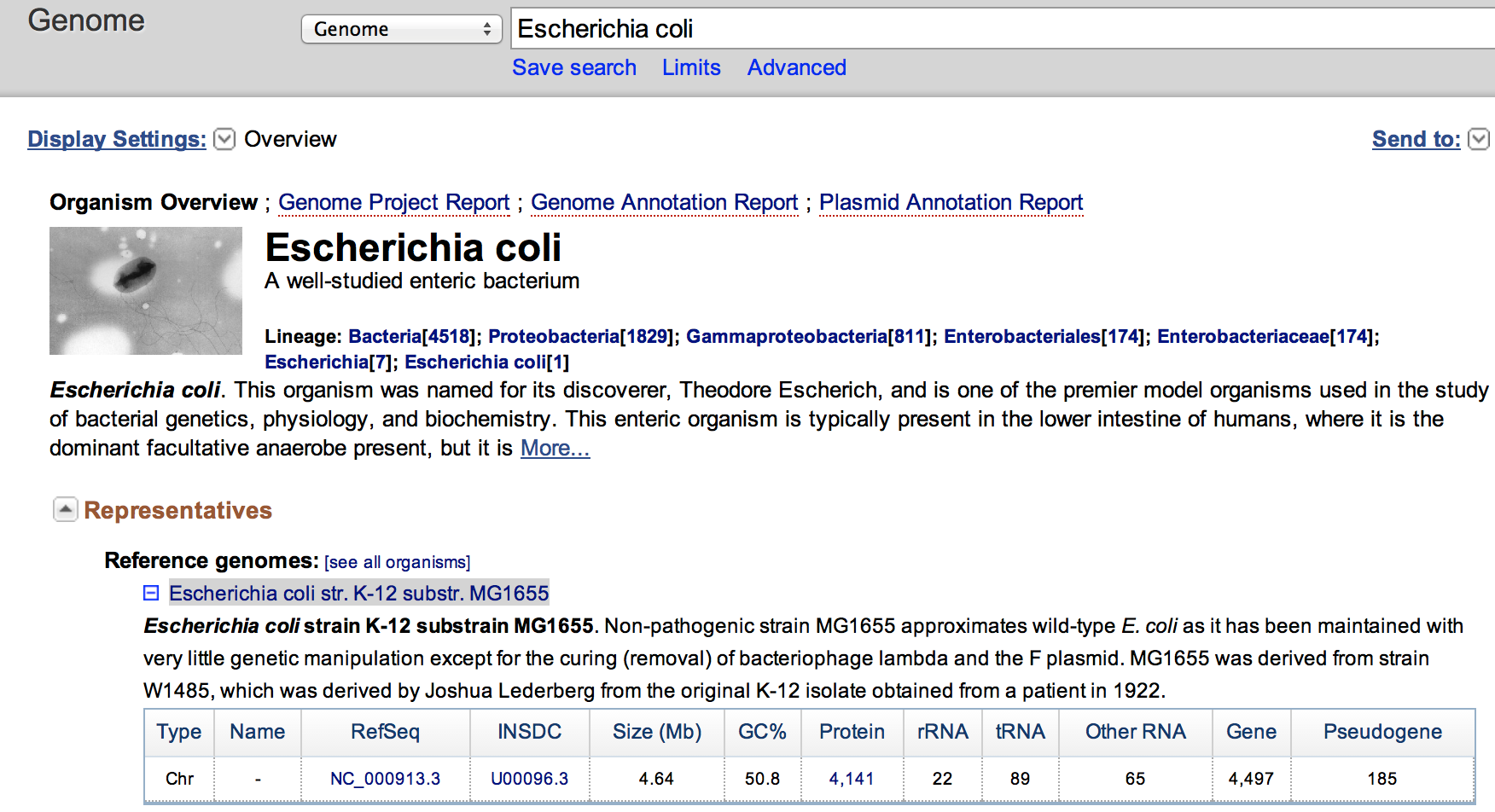
The genome is 4.64 Mbase. The file has 3.6 M reads. As we aim for 10 M for 3 Gb when working with human, the dataset here should enough reads for proper analysis.
At this point, you should be confident about the quality of the datasets, and wether it's worth following with analyzing the datasets.